


Días pasados, un equipo quirúrgico del Hospital Garrahan de Buenos Aires operó con éxito a una niña británica afectada del Síndrome de Holt-Oram, una rara enfermedad congénita que produce malformaciones cardíacas y del miembro superior. La patología de la pequeña implicaba una grave deformidad de ambos dedos pulgares, que le impedía el uso de los mismos. Como la mano humana es, ni más ni menos, una pinza de dos ramas (el pulgar es una, y los restantes dedos la otra), la falta de una de ellas destroza la funcionalidad de la mano. La niña, británica de madre argentina, fácil es imaginarlo, nunca había podido dibujar, alimentarse por sí misma ni siquiera tomar sus juguetes.
La solución del problema —en el caso del defecto en el pulgar— es, como es obvio, quirúrgica, y la operación sólo puede realizarse antes de que el infante cumpla los dos años de edad. La salud pública británica se negaba a operar a la nena, por lo que, ante el inminente cumplimiento del plazo, la madre decidió traerla a su país de origen y buscar ayuda en el Garrahan.
Por supuesto, la encontró.
La mano humana, íntegra y perfecta en sí misma, no es un tema menor: una escuela completa de científicos evolucionistas sostiene que el impactante desarrollo del cerebro humano, circunstancia que nos ha llevado a la posición de especie dominante en este planeta y aledaños, se debió, principalmente, a la necesidad de contar con un sistema nervioso central capaz de controlar y dominar nuestros increíbles pulgares oponibles y su funcionalidad de pinza, única entre todas las especies de la Tierra.
Ni siquiera el famoso "pulgar del panda" puede compararse con el nuestro, ya que el del oso chino no es un verdadero pulgar, ni aún un dedo, sino sólo un sobredimensionado hueso sesamoideo.
Por tanto, asumimos que el tamaño y la complejidad de nuestros cerebros se debieron a la funcionalidad de las manos de nuestros antepasados homínidos, que exigió la creación de nuevas conexiones neuronales para manejar un órgano de tan elevada potencialidad. De este modo, la mano hizo evolucionar al cerebro, y éste, mejorado, a su vez, permitió a la mano realizar tareas cada vez más exquisitas e intrincadamente elaboradas. El Hombre entró así en un "círculo virtuoso" evolución de la mano à evolución del cerebro à evolución de la mano, que nos ha convertido en lo que somos actualmente.
Y todo gracias a la oponibilidad de nuestro pulgar.
Pero la protagonista de esta historia, tristemente, no tenía pulgares funcionales. La pinza carecía de una rama. Había que resolverlo de algún modo, y así se hizo.
El Síndrome de Holt-Oram (SHO por sus siglas en castellano y HOS por las inglesas), ha recibido también los nombres de Displasia Atriodigital, Síndrome del Corazón y la Mano tipo Holt-Oram, y Síndrome Cardíaco y del Miembro Superior. Se trata de una rara anormalidad genética que se caracteriza por acarrear malformaciones congénitas de los huesos del pulgar u otros del miembro superior, así como diversas manifestaciones cardíacas.
Las manifestaciones óseas del HOS (que siempre están presentes) son altamente variables de un caso a otro: la mayor parte de los niños afectados por esta enfermedad presentan pulgares ausentes o hipoplásicos (subdesarrollados). Se observan comúnmente pulgares no funcionales con tres falanges (el pulgar, a diferencia de los demás dedos, sólo tiene dos), así como fusión de varios huesos o desarrollo anormal de otros. También son comunes los huesos supernumerarios en las muñecas (por ejemplo el escafoides), malformaciones en los metacarpianos, hipoplasias del cúbito y el radio, del húmero, los omóplatos y/o las clavículas. Estas fallas pueden ser unilaterales o bilaterales y simétricas o no. En algunos casos, las anormalidades óseas son tan graves que se han visto HOSs focomélicos, esto es, con ausencia total de antebrazos y brazos. En la focomelia, por tanto, las manos salen directamente del tronco. El parecido de este cuadro con el producido por la talidomida hizo que fuese conocido como Síndrome Paratalidomídico. Como se puede observar, las anormalidades pueden ser muchas y muy diversas, y de muy distintos grados de gravedad. Sin embargo, las malformaciones de los huesos del carpo son las únicas que están presentes siempre, en todos los casos de HOS. En otras palabras, si los huesos del carpo no están afectados, el paciente no sufre de HOS. Algunas de estas fallas del desarrollo óseo son tan pequeñas y leves que no afectan la funcionalidad de la mano, y sólo se descubren radiográficamente.
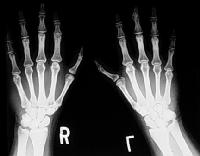
Síndrome de Holt-Oram: la última falange del pulgar izquierdo es hipoplásica,
los huesos del carpo son anormales en ambas manos,
pero la deformidad es más grave del lado izquierdo,
como es habitual en esta enfermedad.
Curiosamente, las malformaciones que afectan al lado izquierdo suelen ser mucho más severas que las localizadas en el brazo o la mano derechos.
La mayoría de los pacientes de HOS (más del 75%) presentan también anormalidades congénitas en el corazón, que oscilan entre pequeñas fallas de conducción eléctrica que son asintomáticas y sólo se evidencian en el electrocardiograma, hasta grandes fallas estructurales que pueden matar al paciente. Así, se ven defectos en el tabique que separa ambas aurículas (defecto septal auricular) o ventrículos (defecto septal ventricular). Las fallas o bloqueos de la conducción eléctrica son más frecuentes que los defectos septales, y suelen aparecer aún en pacientes donde éstos están ausentes.
Mas, ¿por qué se produce esta patología?
El HOS es producido por un gen anormal, ubicado en uno de los autosomas (cromosomas no sexuales). Las estadísticas dicen que, a nivel mundial, el 60% de los pacientes de HOS han heredado el gen de uno de sus padres, también afectado. El restante 40% obedece a mutaciones de novo, esto es, espontáneas. En los Estados Unidos esta relación cambia: el 85% de los casos no tienen un ancestro afectado por el HOS, por lo que se los atribuye a mutaciones espontáneas. Investigada la frecuencia en el mismo país, se averiguó que este síndrome afecta, por suerte, a menos de 1 niño por cada 100.000 nacimientos. Holt-Oram afecta a ambos sexos casi por igual, con cierta preferencia por el sexo femenino, y normalmente mayor severidad en éste que en los varones.
Dada la presencia de antecedentes familiares en algunos de los pacientes afectados de HOS, es importante aclarar que aún las víctimas de mutaciones de novo son capaces, en teoría, de transmitir la enfermedad a su descendencia. En los Estados Unidos, la posibilidad de que el portador de una mutación espontánea herede el HOS a sus hijos es superior al 50%.
La repetición de la misma malformación en miembros de una misma familia llamó la atención de Holt y Oram, quienes, en 1960, estudiaron y describieron esta condición en miembros de cuatro generaciones de una familia. Todos los pacientes, de bisabuelos hasta bisnietos, pasando por abuelos y padres, presentaban defectos septales auriculares y malformaciones en los pulgares.
Profundizando en las causas (etiología) del HOS, se descubrió que depende de mutaciones de un gen, el T-box 5 (TBX5), responsable del desarrollo del corazón y los miembros superiores en la etapa fetal. No se conocen factores ambientales que puedan producir estas mutaciones. Se ha descubierto que las mismas consisten en: sustituciones de nucleótidos que son ilegibles para la transcripción (8 variaciones descubiertas), borrado de pequeños fragmentos del gen (3 variedades), inserciones de fragmentos incorrectos (2 variedades distintas), una mutación que es una combinación de inserción de un fragmento sin sentido y borrado de otro correcto, y una translocación de una secuencia, que puede incluir inversiones. Una o varias de estas fallas, combinadas, producen los muy diversos y extraños tipos de malformaciones que se observan en este síndrome.
El gen defectuoso se encuentra localizado en el brazo largo del cromosoma 12, y la mutación causa la enfermedad desactivando el TBX5. Desafortunadamente, por más que sabemos de la importancia del factor TBX5 en la génesis de los tabiques cardíacos y en el desarrollo del miembro superior, su papel exacto en el proceso todavía no ha sido descubierto. Hatcher y Kim, de la Universidad de Cornell, opinan que el TBX5 participa de la modulación del crecimiento de los tejidos cardíacos embrionarios, pero no mencionan su función en el desarrollo del miembro superior.
Modelo molecular del TBX5
Nuevos estudios y posteriores descubrimientos acerca del papel de TBX5 nos darán, a no dudarlo, un mayor control (y acaso cura intrauterina o terapia genética temprana) sobre esta malhadada enfermedad.
Como quiera que sea, el paciente de Holt-Oram requiere, en muchos casos, de ayuda cardiológica y también quirúrgica (en los casos en que la misma es posible), para restaurar la función del miembro superior. La niña británica operada hace poco en Buenos Aires, afortunadamente, presentaba una malformación carpiana y de los pulgares que, si bien no era recuperable per se, sí admitía la técnica quirúrgica conocida como "pulgarización de los índices".
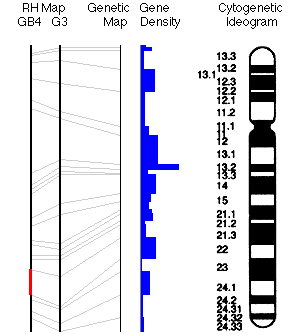
El cromosoma 12. El sector donde se encuentra ubicado
el TBX5 se halla marcado en rojo
La operación, de uso común en personas que han sufrido la amputación de los pulgares, consiste en desplazar el dedo índice, cambiando su ubicación, ángulo y plano, para obligarlo a cumplir las funciones de un pulgar. De este modo, la función trascendente de "pinza" digital, que normalmente realizaría el pulgar, pasa a ser operada por el índice contra los demás dedos. La técnica de pulgarización de los índices es utilizada con frecuencia, es técnicamente simple, casi nunca falla, y tiene la ventaja de no afectar la circulación sanguínea de la mano. Como desventajas, la pinza digital tiene un menor rendimiento (aunque aún se comporta como pinza) y el ancho de la mano queda disminuido. Los lectores interesados pueden ver una descripción técnica de la operación y un descriptivo diagrama en http://cirugia-plastica.org/ (personas impresionables abstenerse).
En la niña de que hablamos, a la que vimos por televisión ya recuperada, la imagen era la de unas pequeñas manitos "de dibujo animado", es decir, de cuatro dedos, pero perfectamente funcionales y cargadas de la ternura de verla dibujar por primera vez en su vida, ya que la ausencia de pulgares funcionales le habían impedido empuñar jamás un instrumento de escritura.
Ante la desconsideración del Estado británico (que se negó a operarla dando múltiples e inadmisibles excusas) hacia esta pequeña víctima, la salud pública argentina reaccionó rápidamente y todo llegó a un final feliz. La pequeña tendrá una vida sana y normal, y el personal del Hospital Garrahan continuará perfeccionándose y mejorando la calidad de vida de los niños.
La protagonista de esta historia con final feliz, entretanto, ya de vuelta en Inglaterra, enfrenta la vida con sus nuevos, flamantes, preciosos índices pulgarizados.
BIBLIOGRAFÍA:
Basson, C.T., Li, R.: "Mutations in TBX5", en http://archive.uwcm.ac.uk/uwcm/mg/search/6175917.html.
Basson, C.T., Huang, T., Lin, R.C. et al.: "Different TBX5 interactions in heart and limb defined by Holt-Oram syndrome mutations", Proc. Natl. Acad. Sci. USA 16 Mar 1999; 96(6): 2919-24.
Basson, Craig T. y Vaughan, Carl J.: "Holt-Oram Syndrome", en http://www.emedicine.com/med/topic2940.htm.
Brockhoff, C.J., Kober, H., Tsilimingas, N., Dapper, F., Myunzel, T.: "Holt-Oram Syndrome". Circulation; 1999 Mar;99(10): 1395-6.
García, Ana, Rosado Diago, José, Palacios Ortega, José: "Manual de cirugía plástica", en http://cirugia-plastica.org/documentos%20manual%2067.html.
Gondres Argote, Rolando, Febles, Daysi, Rondón, Osana et al.: "Síndrome de Holt y Oram. Presentación de un caso", Rev Cubana Ortop Traumatol 2000;14(1-2):56-61
Hatcher, C.J., Kim, M.S. et al.: "TBX5 transcription factor regulates cell proliferation during cardiogenesis", en http://www.ncbi.nlm.nih.gov/entrez/
Radiografía reproducida con permiso de eMedicine.com, Inc., 2003 - Radiographic image reprinted with permission from eMedicine.com, Inc., 2003.